過渡金屬催化有機(jī)反應(yīng)是現(xiàn)代有機(jī)合成化學(xué)中的重要組成,幾十年來得到了化學(xué)家們的密切的關(guān)注,取得了許多令人矚目的成果。其中,Ru催化的烯烴復(fù)分解反應(yīng)、Pd催化的交叉偶聯(lián)反應(yīng)分別獲得2005年、2010年的Nobel化學(xué)獎(jiǎng)。然而,過渡金屬催化劑主要使用的是陸地上稀有的第二行和第三行過渡金屬。近年來,我院王晨副教授與新加坡南洋理工大學(xué)和日本東北大學(xué)的Yoshikai教授課題組展開合作,以地球上富含的廉價(jià)3d過渡金屬Co為催化劑,應(yīng)用于炔烴的功能化反應(yīng),取得了一系列研究成果,在Journal of the American Chemical Society、Angewandte Chemie International Edition、ACS catalysis等國際頂級權(quán)威期刊上發(fā)表多篇高水平學(xué)術(shù)論文。
2020年6月,研究團(tuán)隊(duì)在Journal of the American Chemical Society雜志上發(fā)表了題為《Cobalt/Lewis Acid Catalysis for Hydrocarbofunctionalization of Alkynes via Cooperative C?H Activation》的文章,發(fā)展了一種由鈷-二膦配合物和路易斯酸(LA,如 AlMe3)組成的催化體系,可以通過實(shí)現(xiàn)對路易斯堿性的缺電子底物(如甲酰胺、吡啶酮、吡啶和相關(guān)嗪、咪唑 [ 1,2-a] 吡啶和唑衍生物)的位點(diǎn)選擇性的C—H 活化,而實(shí)現(xiàn)對炔烴的氫羰基功能化。與已知的用于類似轉(zhuǎn)化的 Ni/LA 催化系統(tǒng)相比,該催化體系的特色在于一方面使用了廉價(jià)且實(shí)驗(yàn)室穩(wěn)定的預(yù)催化劑和配體如 Co(acac)3和1,3-雙(二苯基膦)丙烷 (dppp),而且對吡啶酮和吡啶衍生物的C—H活化顯示出與Ni催化體系不同的位點(diǎn)選擇性。特別是首次實(shí)現(xiàn)了吡啶的完全 C4 選擇性烯基化。同時(shí),該催化系統(tǒng)還可以選擇性的對咪唑并 [1,2-a] 吡啶衍生物實(shí)現(xiàn)C5 選擇性烯基化。通過DFT計(jì)算對 Co/ AlMe3催化的N,N-二甲基甲酰胺對炔烴的加成反應(yīng)機(jī)理研究表明,該反應(yīng)經(jīng)歷配體到配體氫轉(zhuǎn)移 (ligand-to-ligand hydrogen transfer, LLHT) 生成烯基(氨基甲酰基)鈷中間體的歷程進(jìn)行,甲酰胺的羰基 C—H鍵的裂解是反應(yīng)的決速步驟。

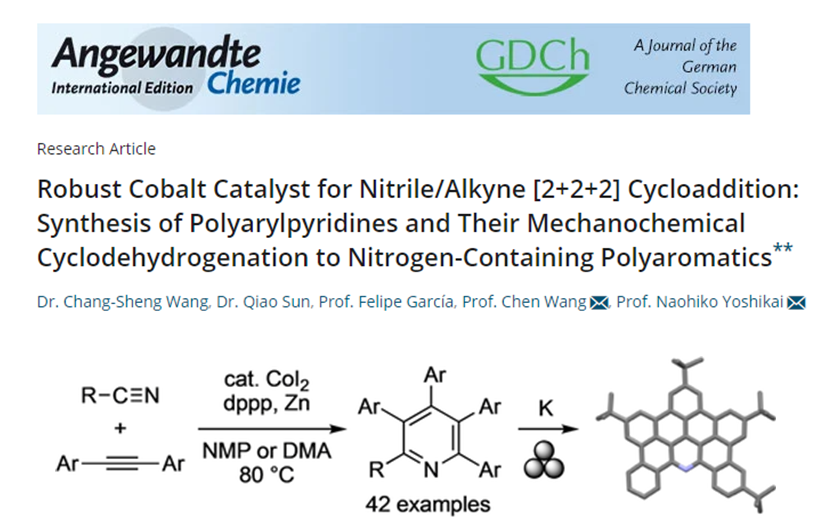
2021年2月,研究團(tuán)隊(duì)在Angewandte Chemie International Edition雜志上發(fā)表了題為《Robust Cobalt Catalyst for Nitrile/Alkyne [2+2+2] Cycloaddition: Synthesis of Polyarylpyridines and Their Mechanochemical Cyclodehydrogenation to Nitrogen-Containing Polyaromatics》的文章,發(fā)展了一種基于碘化鈷(II),1,3-雙(二苯基膦基)丙烷和鋅的簡單廉價(jià)的催化體系,可促進(jìn)各種腈和二芳基乙炔的[2+2+2]環(huán)加成反應(yīng),從而合成一系列多芳基吡啶。DFT機(jī)理研究表明反應(yīng)依次經(jīng)歷兩個(gè)炔烴氧化偶聯(lián)、腈插入鈷環(huán)戊二烯中間體和C‐N還原消除。反應(yīng)獲得的四芳基和五芳基吡啶通過機(jī)械化學(xué)輔助的還原性脫氫環(huán)化,可以合成全新的含氮多環(huán)芳烴。
2022年3月,研究團(tuán)隊(duì)在ACS catalysis雜志上發(fā)表了題為《Cobalt-Catalyzed Carbo- and Hydrocyanation of Alkynes via C?CN Bond Activation》的文章,發(fā)展了鈷催化的通過有機(jī)氰化物的 C-CN 鍵斷裂發(fā)生的炔烴的碳氰基化和氫氰基化反應(yīng)。研究發(fā)現(xiàn),通過用 Zn 還原生成的低價(jià)鈷-二膦催化劑可以實(shí)現(xiàn)反式選擇性的炔烴的芳基氰基化。相比之下,向該催化體系中添加 Zn(OTf) 2不僅可以加速轉(zhuǎn)化,而且改變了立體選擇性,最后得到順式的芳基氰化產(chǎn)物。該催化體系對于使用烯基氰化物的烯基氰化以及使用丙腈作為 HCN 替代物的轉(zhuǎn)移氫氰基化都非常行之有效。機(jī)理實(shí)驗(yàn)揭示了生成活性鈷催化劑的長誘導(dǎo)期、立體選擇性的動(dòng)力學(xué)控制以及芳基-CN鍵活化步驟的性質(zhì)。DFT計(jì)算對芳基氰化的機(jī)理研究表明,反應(yīng)歷程包含了芳基?CN的氧化加成、炔烴插入Co?芳基鍵和烯基?CN還原消除。其中烯基(氰基)鈷中間體的順式/反式異構(gòu)化可能先于還原消除發(fā)生。在Co-dppp體系中,順式/反式異構(gòu)化較容易發(fā)生,和順式和反式產(chǎn)物的還原消除途徑相互競爭。而Zn(OTf)2的加入則可以通過穩(wěn)定過渡態(tài)降低C?CN還原消除的活化能,從而使順式芳基氰化途徑更加有利。

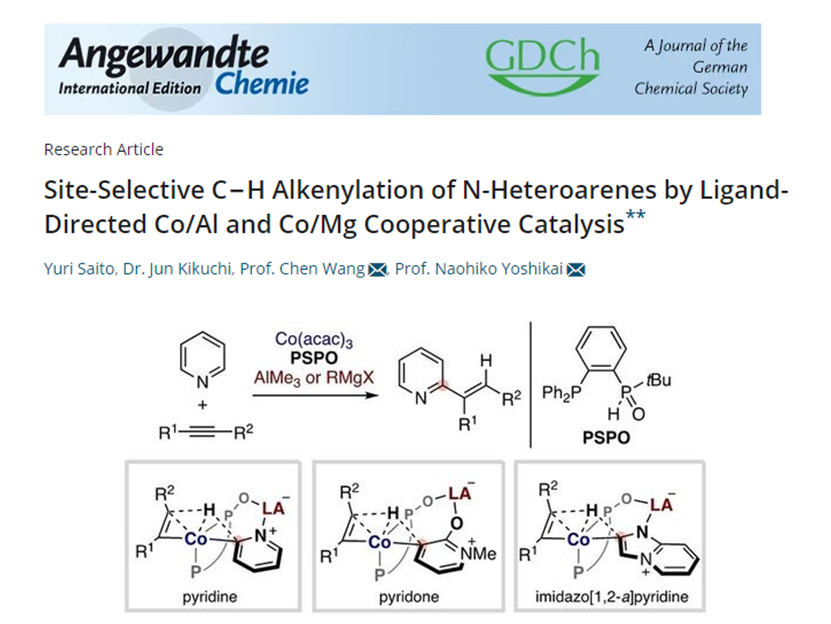
2023年3月,研究團(tuán)隊(duì)在Angewandte Chemie International Edition雜志上發(fā)表了題為《Site-Selective C?H Alkenylation of N-Heteroarenes by Ligand-Directed Co/Al and Co/Mg Cooperative Catalysis》的文章,發(fā)展了使用Co/Al和Co/Mg雙金屬催化劑和膦/仲氧化膦雙功能配體(PSPO),催化 N-雜芳烴與炔烴的位點(diǎn)選擇性 C-H 烯基化反應(yīng)。吡啶、吡啶酮和咪唑并 [1,2- a ]吡啶在靠近路易斯堿性氮或氧的C-H位點(diǎn)都能被有效地烯基化。DFT計(jì)算表明C-H活化歷程是配體到配體氫轉(zhuǎn)移 (ligand-to-ligand hydrogen transfer, LLHT)。
王晨副教授是上述系列論文的共同通訊作者,負(fù)責(zé)課題中的DFT計(jì)算部分,我校是論文[1]、[2]、[4]的第二單位,論文[3]的第四單位。部分工作得到了浙江省自然科學(xué)基金(LY18B020007)的支持。
近期發(fā)表的相關(guān)文章:
[1]Cobalt/Lewis Acid Catalysis for Hydrocarbofunctionalization of Alkynes via Cooperative C?H Activation. J. Am. Chem. Soc. 2020, 42, 12878-12889.
論文鏈接:https://pubs.acs.org/doi/10.1021/jacs.0c06412
[2]Robust Cobalt Catalyst for Nitrile/Alkyne [2+2+2] Cycloaddition: Synthesis of Polyarylpyridines and Their Mechanochemical Cyclodehydrogenation to Nitrogen-Containing Polyaromatics. Angew. Chem. Int. Ed. 2021, 60, 9627-9634.
論文鏈接:https://doi.org/10.1002/anie.202017220
[3]Cobalt-Catalyzed Carbo- and Hydrocyanation of Alkynes via C?CN Bond Activation. ACS Catal. 2022, 12, 4054?4066.
論文鏈接:https://doi.org/10.1021/acscatal.2c00181
[4]Site-Selective C?H Alkenylation of N-Heteroarenes by Ligand-Directed Co/Al and Co/Mg Cooperative Catalysis. Angew. Chem. Int. Ed. 2023, 62, e202301006.
論文鏈接:https://doi.org/10.1002/anie.202301006
作者簡介:
王晨,現(xiàn)為紹興文理學(xué)院化學(xué)化工學(xué)院副教授。2010年12月畢業(yè)于中國科學(xué)技術(shù)大學(xué),獲得理學(xué)博士學(xué)位。主要從事的研究方向是理論與計(jì)算有機(jī)化學(xué),已發(fā)表SCI論文20多篇,主持國家和省級項(xiàng)目各一項(xiàng)。
文字:廖慶 編輯:嚴(yán)許媖
 教職工
教職工

 學(xué)生
學(xué)生
 考生
考生
 VPN
VPN
 辦事大廳
辦事大廳 電子郵箱
電子郵箱 新聞網(wǎng)
新聞網(wǎng) English
English 返回舊版
返回舊版




 浙公網(wǎng)安備 33030402000759號
浙公網(wǎng)安備 33030402000759號

